Contents
- Overview
- Installation
- Dependencies
- Commands and options
- Finding signatures of genes or transcripts
- Applying signatures of genes or transcripts in survival
- Integrative analysis
- Toy example
- Authors
Overview
Reboot is a flexible, easy-to-use algorithm to identify and validate genes or transcripts signatures whose expression are highly correlated with patient survival. This tool innovates by using a multivariate strategy with penalized Cox regression (Lasso method) combined with a bootstrap approach, presenting robust convergence of the regression procedure, and a variety of statistical tests for signature score. Reboot comprises two modules developed in R (version 4.0.4). The regression module provides functionality for obtaining gene/transcript signatures from a given set of samples. In turn, the survival module provides functionality for producing, applying and validating a score, which is calculated based on the obtained signature, in patient datasets. In this module, a different set of samples may be provided for validation purposes, and clinical variables may also be taken into account. Moreover, Reboot also has the execution option complete, which integratively executes the two aforementioned modules.
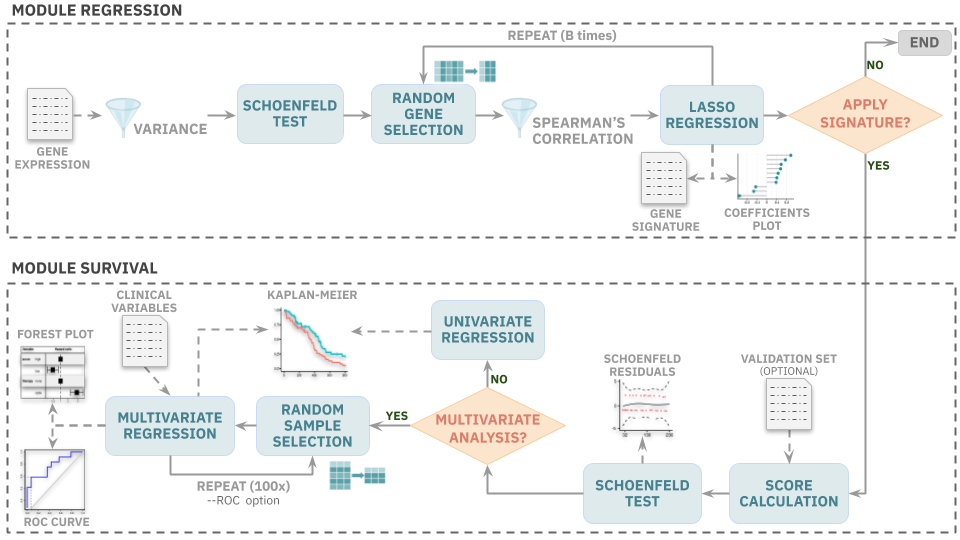 Reboot workflow: First module (regression) makes a regression analysis to identify a gene or transcript signature. The second module (survival) runs survival analysis of a score calculated based on the obtained signature.
Reboot workflow: First module (regression) makes a regression analysis to identify a gene or transcript signature. The second module (survival) runs survival analysis of a score calculated based on the obtained signature.
Installation
Reboot can be obtained from Github and installed via a Docker container (recommended) or through direct installation.
-
Docker container
This method works on any distribution or operational systems, as long as Docker is installed.
docker pull galantelab/reboot -
Direct installation
This method requires the previous installation of R (version >= 4.0.4):
git clone https://github.com/galantelab/reboot.gitsudo sh reboot/install.sh
Dependencies
In order to work properly, the following packages are necessary (included in the installation procedures):
- argparse
- BiocManager
- BioinformaticsFMRP/TCGAbiolinks (from BiocManager)
- data.table
- extrafont
- forestmodel
- hash
- OptimalCutpoints
- optparse
- penalized
- remotes (from BiocManager)
- R.utils
- sjlabelled
- sjmisc
- sjstats
- survcomp (from BiocManager)
- survival
- survivalROC
- survminer
- tidyverse
- mice
Commands and options
Reboot works with a command and subcommands structure:
reboot.R [subcommand] <options>
Subcommands may be invoked by the help menu:
# for docker container
docker run --rm galantelab/reboot reboot.R -h
Optionally:
# for direct installation
reboot.R -h
Similarly, version may be invoked by:
# for docker container
docker run --rm galantelab/reboot reboot.R -v
Optionally:
# for direct installation
reboot.R -v
In summary, 3 subcommands are available:
| regression | generates a signature through multivariate Cox regression |
| survival | applies a signature score in survival analysis |
| complete | generates a signature and applies its derived score in survival analysis |
Finding signatures of genes or transcripts
Reboot searches in the regression module for a genetic signature (significance coefficients) correlated with patient survival based on a multivariate Cox regression of genes or transcripts. This module uses a LASSO algorithm combined with a bootstrap approach to deal with possible dimension vulnerabilities (especially when data has many attributes and few instances).
In addition, a number of filters are implemented. Reboot analyses starts off by checking if the provided expression dataset has the minimum of 20 variables (genes/transcripts) necessary for the next steps to take place correctly. Also, a minimum of 10 samples is required due to cross validation steps performed to optimize LASSO coefficient choice. Then, data attributes with variance lower than a user-defined cut-off are removed. If a low variance is detected in the provided follow up times or the proportion between individuals with diverging survival status (e.g., dead and alive) is greater than 80% or lower than 20%, the analysis halts and returns a warning due to possible convergence problems in the regression process. Prior to starting the analysis, a Schoenfeld’s test filter is also performed in a univariate way to exclude variables which do not meet the proportional hazard assumptions of Cox models. Next, a Spearman’s correlation filter is applied for every iteration of the bootstrap process based on the user-defined allowed fraction of pairs with correlation coefficients higher than 0.8 and p-values lower than 0.05. To minimize false positives in the results, empirical coefficient cutoffs for genes/transcripts were established: genes with coefficients higher than 0.0035 are selected, whereas only transcripts with coefficients higher than 0.011 are chosen based on random bootstrap resampling analyses.
Coverage (Cv) may be introduced as the number of times an attribute is raffled, on average, after B iterations, according to the formula:
Cv = (B * G) / N
Where (N) is the number of attributes (genes or transcripts) and (G) is the group size, i.e, the number of attributes to be included in each iteration. For optimal algorithm convergence, we recommend the group size (G) to be between 10 and 15 attributes per iteration. In order to assure a satisfying level of group combinations, our suggestion is to run each attribute in N/G simulations on average. In other words: Cv = N/G, which yields:
B = (N / G)²
For instance, when (N) is 100 and (G) is 10, the recommended number of bootstrap iterations would be 100 and, as a consequence, the number of appearances of each attribute would be 10, as exemplified in the following table:
| N | G | B | Cv |
|---|---|---|---|
| 100 | 10 | 100 | 10 |
| 300 | 15 | 400 | 20 |
| 500 | 12 | 1736 | 42 |
We recommend avoidance of regression set that may not present unimodal distribution, which may introduce some bias. The regression analysis implemented in Reboot is performed approximately linearly in time in relation to the number of bootstrap iterations. If the analysis should take no longer than a period of the day, we encourage a maximum of 10,000 iterations.
Usage
For gene or transcript signature obtention, run the following:
# for docker container
docker run --rm -v $(pwd):$(pwd) galantelab/reboot reboot.R regression <options>
optionally:
# for direct installation
reboot.R regression <options>
Regression options are:
| Options | Description |
| -I, --filein | Input file name. Tab-separated values (.tsv) file containing genes or transcripts expression and survival parameters |
| -O, --outprefix | Output file prefix (string). Default: reboot |
| -B, --bootstrap | Number of iterations for bootstrap simulation (integer). Default: 1 |
| -G, --groupsize | Number of genes or transcripts to be selected in each bootstrap simulation (integer). Default: 10 |
| -P, --pcentfilter | Percentage of correlated gene or transcript pairs allowed in each iteration (double). Default: 0.3 |
| -V, --varfilter | Minimum normalized variance (0-1) required for each gene or transcript among samples (double). Default: 0.01 |
| -T, --ty | Type of transcriptome data to be analyzed: gene or transcript (character). Default: gene |
| -F, --force | Choose -F to bypass OS and OStime filters |
| -h, --help | Show this help message and exit |
Input
To produce a genetic signature, Reboot requires a .tsv file containing normalized expression values in Transcripts Per Million (TPM) or in Fragments Per Kilobase per Million (FPKM) for genes or transcripts across multiple samples, in addition to survival data: survival status (e.g., 0 = alive or 1 = dead) and follow up time:
| Sample ID | OS | OS.time | PARPBP | RAD51 | … |
|---|---|---|---|---|---|
| patient_1 | 1 | 448 | 41.81557 | 34.70869 | … |
| patient_2 | 0 | 466 | 24.78227 | 64.80153 | … |
| … | … | … | … | … | … |
Output
As result, Reboot generates a log file, a .txt file containing mean regression coefficients (with standard deviation, where applicable) for the signature and 2 plots in .pdf. The signature.txt file is in the following format:
| feature | coefficient | sd |
|---|---|---|
| PARPBP | 0.17014 | 3.13942 |
| CXCR6 | 0.22173 | 2.16845 |
| … | … | … |
Coefficients may be interpreted according to absolute value and signal. Significance is as high as the absolute value that the coefficient gets. Positive signals contribute to the accountability of bad prognosis, while negative signals contribute to the accountability of good prognosis.
The plots generated are a histogram with the distribution of the regression coefficients and a lollipop plot with the most relevant coefficients (see bellow).

Applying signatures of genes or transcripts in survival
Reboot produces and applies in the survival module a score for all samples based on the signature previously obtained from the regression module according to the following formula:
Where “C” is the Cox regression coefficient and “E” is the expression value of each gene/transcript in the signature.
Besides, Reboot also offers the multivariate option, where further clinical variables (e.g., therapy, age and gender) can be loaded in a multivariate survival model. Multiple univariate analyses are executed and only variables with a p-value <= 0.2 and that passed the Schoenfeld’s test are selected for the final multivariate model. Statistical tests are performed in order to evaluate the relevance of the signature score along with co-variables as prognostic factors of a given event (overall / progression-free / recurrence-free survival).
By default, both univariate and multivariate survival analyses use the median score value as a cutoff to stratify patients in high and low score signatures. Alternatively, this cutoff value may be based on a Receiver Operator Characteristic (ROC) curve using Nearest Neighbour Estimate (NNE) method and the Youden statistics, where J = [sensitivity + (specificity -1)]. If more than one J coefficient is available, then the first one is chosen.
If a multivariate analysis is performed based on a ROC curve, a bootstrap resampling method is applied once the provided clinical dataset passes the following additional filters: (i) final dataset with at least 70% of the original one and; (ii) the frequency of the less abundant category for each co-variable is not less than 20% (proportion filter). Otherwise, the multivariate analysis is performed without the bootstrap method. After 100 iterations, the relevance frequency of each co-variable with the event is calculated.
Usage
To validate a signature of genes or transcripts in survival analysis, run the following:
# for docker container
docker run --rm -v $(pwd):$(pwd) galantelab/reboot reboot.R survival <options>
Optionally:
# for direct installation
reboot.R survival <options>
Survival options are:
| Options | Description |
| -I, --filein | Input file name. Tab-separated values (.tsv) file containing genes or transcripts expression and survival parameters |
| -O, --outprefix | Output file prefix (string). Default: reboot |
| -S, --signature | Tab-separated values (.tsv) file containing a set of genes or transcripts and corresponding cox coefficients |
| -M, --multivariate | If clinical variables should be included, choose -M. This option is tied with -C option. Default: FALSE |
| -C, --clinical | Tab-separated values (.tsv) file containing binary categorical variables only. Required if -M option is chosen |
| -R, --roc | To categorize the genetic score according to a ROC curve instead of median value, choose -R. Default: FALSE |
| -V, --varfilter | Minimum normalized variance (0-1) required for follow up time (double). Default: 0.01 |
| -F, --force | Choose -F to bypass OS and OStime filters |
| -h, --help | Show this help message and exit |
Inputs
Survival analyses may be run in univariate or multivariate mode. Required inputs depend on this choice.
-
Univariate mode
This is the simplest mode and requires a single input file. The expected .txt file contains a set of features (genes/transcripts) and their corresponding coefficients provided as output by the regression module:
feature coefficient PARPBP 0.17014 CXCR6 0.22173 … … -
Multivariate mode
In case multivariate mode is chosen, a .tsv file containing clinical information is also necessary. Note that all clinical variables must be categorical and present only 2 classes (NA values are allowed):
Sample ID age gender therapy … patient_1 18-55 years male radiation … patient_2 56+ years female chemoradiation … … … … … …
Outputs
Depending on whether survival analysis was performed in univariate or multivariate mode, a different set of output files are created.
-
Univariate mode
If the analysis is performed in univariate mode, Reboot returns 4 files: (i) log; (ii) univariate summary results; (iii) table with the scores calculated for all patients; (iv) and an updated signature file for interpretation.
The uniCox.txt file contains the survival results for the signature score:
feature control condition coefficient hazard.ratio log.rank.pvalue low.high.samples median.survival.low median.survival.high prognosis score high low -1.0091 0.3645 (95% CI, 0.2456-0.541) 0.003 52/53 532 (95% CI, 455-648) 313 (95% CI, 231-362) better The scoreCont_table.tsv file contains the scores calculated for each patient:
OS OS.time score patient_1 0 518 -0.0015 patient_2 0 2022 -0.1929 patient_3 1 395 0.1239 ... ... ... ... The signature_updated.tsv file contains the same features and coefficients from the regression module, with an additional column translating the values into prognosis:
feature coefficient sd prognostic PARPBP 0.17014 3.13942 worse CXCR6 0.22173 2.16845 worse NTRK1 -0.1517 1.84659 better ... ... ... ... Plots in .pdf returned in this mode include: a proportional hazard assumptions plot (result of Schoenfeld test) and a Kaplan Meier plot (see bellow).
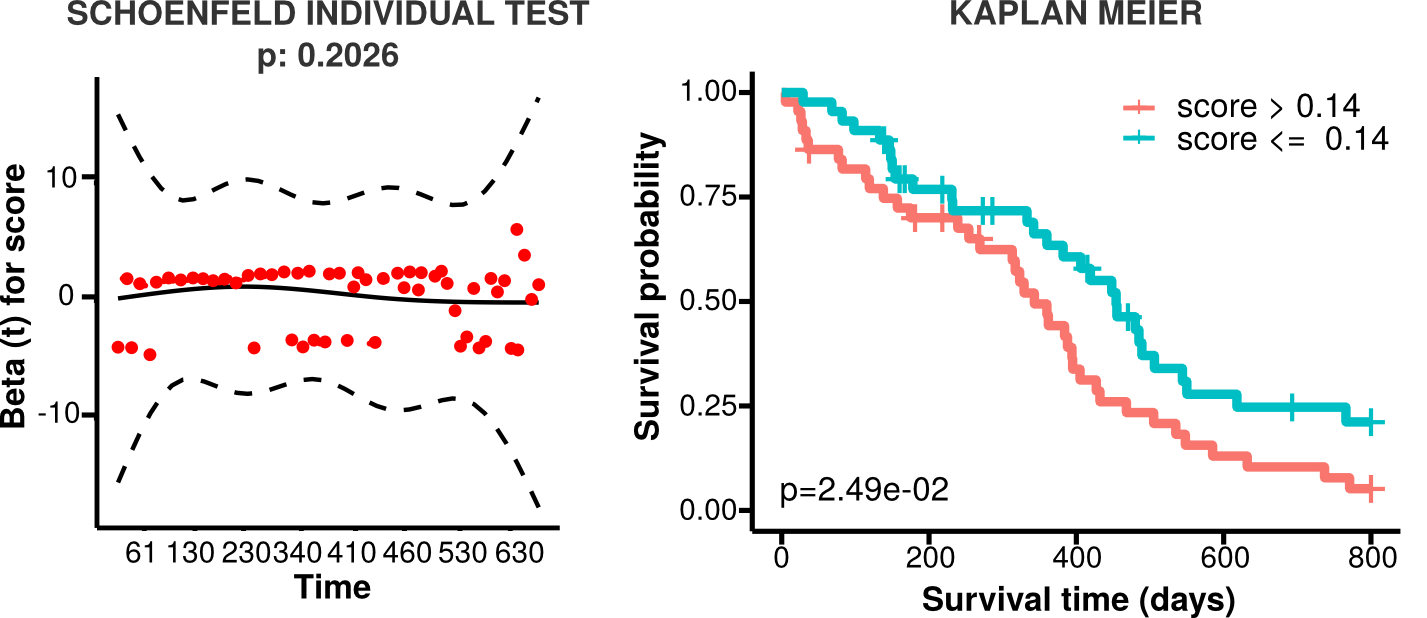
-
Multivariate mode
If the analysis is performed in multivariate mode, Reboot returns all files created in the univariate mode in addition to a multicox.txt file containing the survival results of the signature score along with all other clinical variables:
variable condition univariate.hazard.ratio univariate.Cox.pvalue univariate.prognosis multivariate.hazard.ratio multivariate.Cox.pvalue multivariate.prognosis score low 0.3645 (95% CI, 0.2456-0.541) 0.001 better 0.3904 (95% CI, 0.2248-0.6779) 8e-04 better age 56+ years 1.369 (95% CI, 0.9086-2.0625) 0.1332 worse 1.1104 (95% CI, 0.6314-1.9531) 0.7161 ---- gender MALE 0.9474 (95% CI, 0.6381-1.4066) 0.7886 ---- ---- ---- ---- ... ... ... ... ... ... ... ... The signature score groups - after the cutoff is applied - and all other clinical variables used in the final multivariate model are displayed in the scoreCat_table.tsv file for further inspection:
OS OS.time score age gender patient_1 0 518 low 18-55 years male patient_2 0 2022 low 18-55 years female patient_3 1 395 high 56+ years male ... ... ... ... ... ... Plots in .pdf returned in this mode include all figures created in the univariate mode in addition to a forest plot for all clinical variables evaluated. If the option –ROC is selected, a ROC curve is also generated (see below). In this case (-R TRUE), a barplot of co-variable frequencies may also be provided for the most relevant variables (p-value <= 0.05 in at least 25% of iterations).
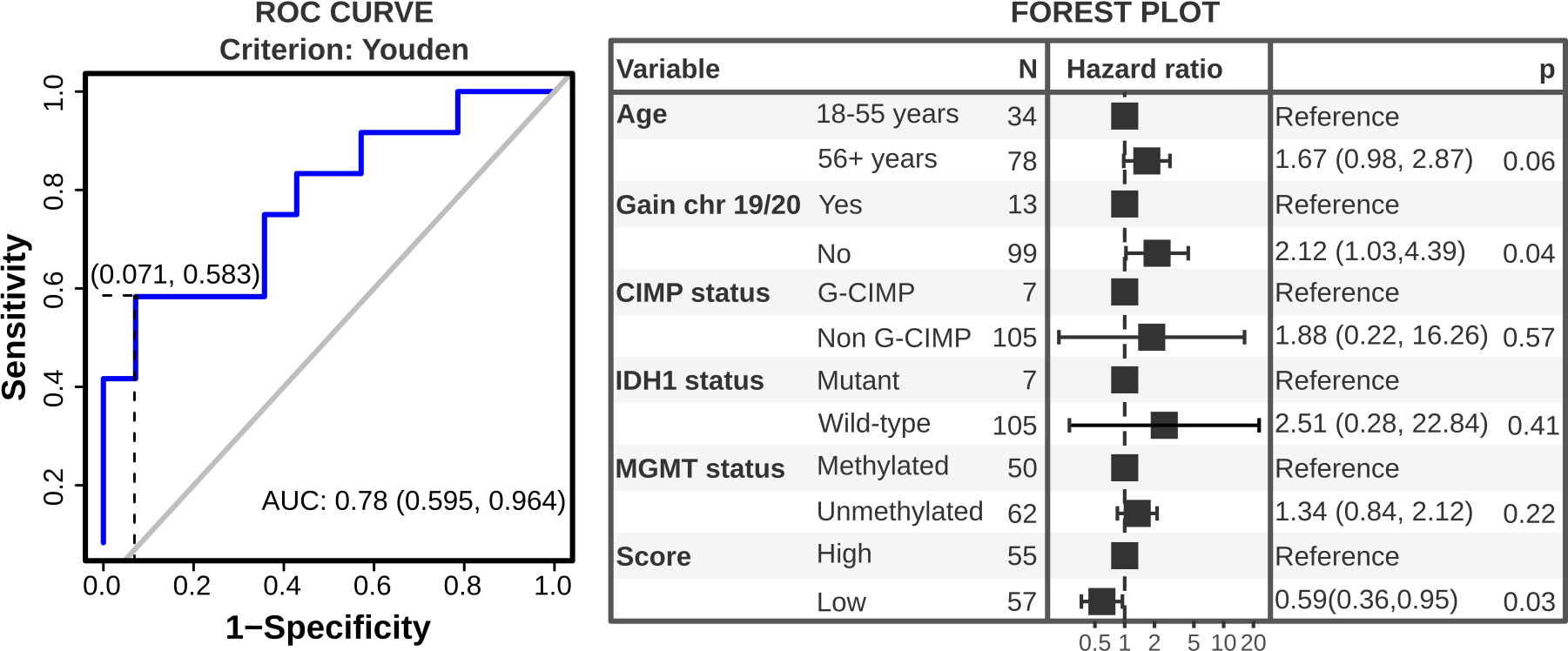
Integrative analysis
Reboot also provides a subcommand to perform the full analyses (regression followed by survival) in a one-step process. To execute it, run the following:
# for docker container
docker run --rm -v $(pwd):$(pwd) galantelab/reboot reboot.R complete <options>
Optionally:
# for direct installation
reboot.R complete -h
Complete options are:
| Options | Description |
| -I, --filein | Input file name. Tab-separated values (.tsv) file containing genes or transcripts expression and survival parameters |
| -O, --outprefix | Output file prefix (string). Default: reboot |
| -B, --bootstrap | Number of iterations for bootstrap simulation (integer). Default: 1 |
| -G, --groupsize | Number of genes or transcripts to be selected in each bootstrap simulation (integer). Default: 10 |
| -P, --pcentfilter | Percentage of correlated gene or transcript pairs allowed in each iteration (double). Default: 0.3 |
| -T, --ty | Type of transcriptome data to be analyzed: gene or transcript (character). Default: gene |
| -V, --varfilter | Minimum normalized variance (0-1) required for each gene or transcript among samples (double). Default: 0.01 |
| -M, --multivariate | If clinical variables should be included, choose -M. This option is tied with -C option. Default: FALSE |
| -C, --clinical | Tab-separated values (.tsv) file containing binary categorical variables only. Required if -M option is chosen |
| -R, --roc | To categorize the genetic score according to a ROC curve instead of median value, choose -R. Default: FALSE |
| -F, --force | Choose -F to bypass OS and OStime filters | -h, --help | Show this help message and exit |
Toy example
In order to illustrate how easy is to use Reboot, we produce a framework to get a small dataset (here called toy dataset) from TCGA and use it into Reboot.
We provide a script to download and format gene expression and clinical data of glioblastoma patients from TCGA. It can be run following this command into the reboot directory:
# for docker container
docker run --env MYID=$(id -u) --rm -ti -v $(pwd):$(pwd) -w $(pwd) galantelab/reboot toyfordocker.R
optionally:
# for direct usage
toyscript.R
This command returns 2 .tsv files, mentioned above, called expression.tsv and clinical.tsv. A MANIFEST.txt file and a set of expression and clinical data are also created, as intermediates of the TCGA download process. The composition of the expression dataset comprises clinical variables: OS (survival status) and OS.time (follow up time) and 50 random picked genes with expression in FPKM.
Finally, Reboot can be run in the complete mode:
# for docker container
docker run -u $(id -u):$(id -g) --rm -v $(pwd):$(pwd) -w $(pwd) galantelab/reboot reboot.R complete -I expression.tsv -O toy -B 100 -G 10 -M -C clinical.tsv -F
Optionally:
# for direct usage
reboot.R complete -I expression.tsv -O toy -B 100 -G 10 -M -C clinical.tsv -F
Authors
Felipe Rodolfo Camargo dos Santos*
Gabriela Der Agopian Guardia*
Filipe Ferreira dos Santos*
Daniel Takatori Ohara
Pedro Alexandre Favoretto Galante
*These authors contributed equally for the work